A Study on Host Tropism Determinants of Influenza Virus using Machine Learning
Current Bioinformatics (2020)
.png)
연구배경
인플루엔자 바이러스는 복제주기의 여러 단계에 걸쳐 숙주 특이적 요소들과 상호 작용하며, 이러한 요소들은 숙주 생물 종에 따라 차이가 있다. 숙주 범위의 변화를 유발하고 특정 숙주 생물들과의 상호 작용 가능성을 증가시키는 인플루엔자 바이러스의 숙주 친화성 결정 요인들은 다양한 숙주 종에서의 바이러스 감염 및 전파를 이해하는 데 중요하다. 본 연구에서는 인플루엔자 바이러스의 숙주 친화성 결정 요인에 대한 생물정보학적 분석을 통해 숙주 예측에 유용한 아미노산 위치 마커 및 물리화학적 특성을 확인하고, 기계학습 알고리즘에 기반한 바이러스의 숙주 분류 및 예측을 통해 미래에 발생 가능한 신종 인플루엔자 바이러스의 감염 특성을 예측하기 위한 시스템을 구축하고자 하였다.
연구방법
세 가지 클래스의 숙주 (조류, 인간, 돼지)로부터 분리된 인플루엔자 바이러스 아형들의 6가지 단백질 HA, NA, PA, PB1, PB2, NP 서열을 수집하여 데이터셋을 구성한 후, 무작위 추출을 통해 선별된 각 단백질별 서열 데이터를 디지털화하고 특징 벡터로 변환하였다. 숙주 분류를 위한 기계학습 기법으로는 랜덤 포레스트(RF), 나이브 베이즈(NB) 및 k-최근접 이웃(kNN) 알고리즘이 사용되었다. 각 기계학습 알고리즘을 적용한 분류의 수행에 따라 각 단백질 서열 데이터 셋별로 숙주 분류에 큰 영향을 미치는 주요한 물리화학적 요인을 탐색하였다. 숙주-특이적 아미노산 위치들을 선별하고 분류 마커로서의 유용성을 확인하기 위해 Java 언어를 사용하여 서열분석 및 숙주별 위치 마커 식별 프로그램을 개발 수행하였다.
연구결과
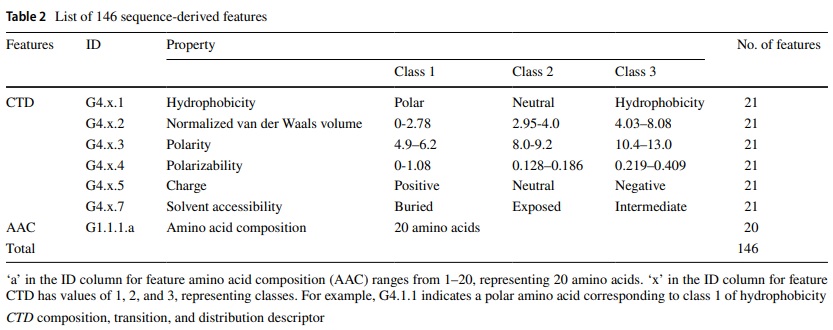
여섯 가지 단백질 유형 중 HA 단백질은 인플루엔자 바이러스의 숙주 친화성을 결정하는데 가장 중요한 역할을 하는 것으로 확인되었으며, 세 가지 알고리즘 중 랜덤 포레스트 기법이 숙주 예측에서 가장 높은 정확도를 나타냈다.
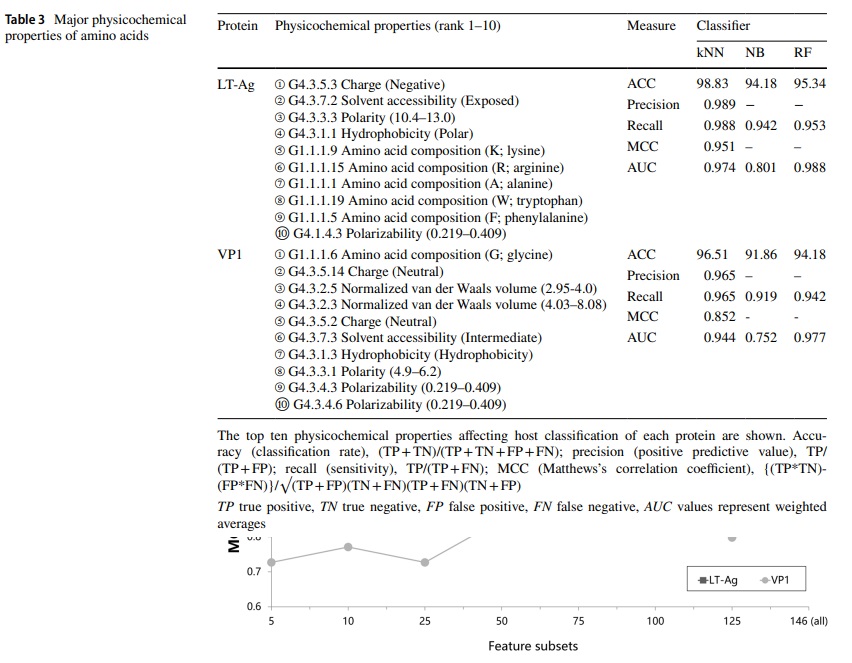
최적의 분류 성능을 보인 랜덤 포레스트 알고리즘에 순위 기반 속성 선택 기법을 적용하여 각 단백질 서열별로 최적의 분류를 위한 특징 집합을 확인함으로써 중요도가 낮은 속성들의 제거를 통한 분류기 성능의 향상 효과를 확인할 수 있었다. 또한 숙주-특이적 차이를 나타내는 보존된 아미노산을 선별 및 확인하고, 이들이 숙주 분류에 유용한 위치 마커임을 제시할 수 있었다. 마지막으로, ANOVA 분석 및 post-hoc 검증을 통해 위치 마커들을 포함하도록 재조합된 단백질 서열들을 구성하는 아미노산의 물리화학적 특성들이 숙주마다 유의하게 상이함을 확인하였다.
연구결론
본 연구에서 사용된 생물정보학적 방법은 숙주-특이적 아미노산 위치 마커와 인플루엔자 바이러스 숙주 종의 식별을 통해 분자 수준에서 바이러스의 숙주 범위 및 감염 특성을 결정 또는 예측할 수 있는 가능성을 시사한다. 연구 결과는 기계학습 기법을 사용한 인플루엔자 바이러스의 숙주 예측 알고리즘 선택 및 적용, 바이러스의 숙주친화성 결정요인의 물리화학적 특성 연구, 위치 마커의 활용을 통한 신·변종 인플루엔자 바이러스의 감염특성 분류, 식별 및 예측 등에 참조 및 활용될 수 있다.