Analysis of protein determinants of host‐specific infection properties of polyomaviruses using machine learning
GENES AND GENOMICS (2021)
.png)
연구배경
대형 종양 항원(large tumor antigen, LT-Ag)과 주요 캡시드 단백질 VP1은 종양 원인 바이러스인 폴리오마바이러스(polyomaviruses, PyV)의 숙주 특이적 감염 특성을 결정하는 데 중요한 역할을 하는 것으로 알려져 있다. 이에 본 연구에서는 PyV 숙주를 예측하기 위해 적용될 수 있는 분류 기술뿐만 아니라 숙주 특이성에 중요한 영향을 미치는 LT-Ag 및 VP1 아미노산의 물리화학적 특성을 조사하기 위한 분석을 수행하였다.
연구방법
분석을 위해 86개의 바이러스 종에 대한 참조 서열을 수집하였다. 재구성된 계통 발생 수의 클러스터링 패턴을 기반으로 하여 수집된 데이터 세트를 포유류, 조류, 어류의 세 그룹으로 나누었고, 숙주 분류를 위해 RF (Random Forest), NB (Naive Bayes) 및 k-최근접 이웃(k-nearest neighbors, kNN) 알고리즘과 같은 머신러닝 기법을 사용하였다.
연구결과
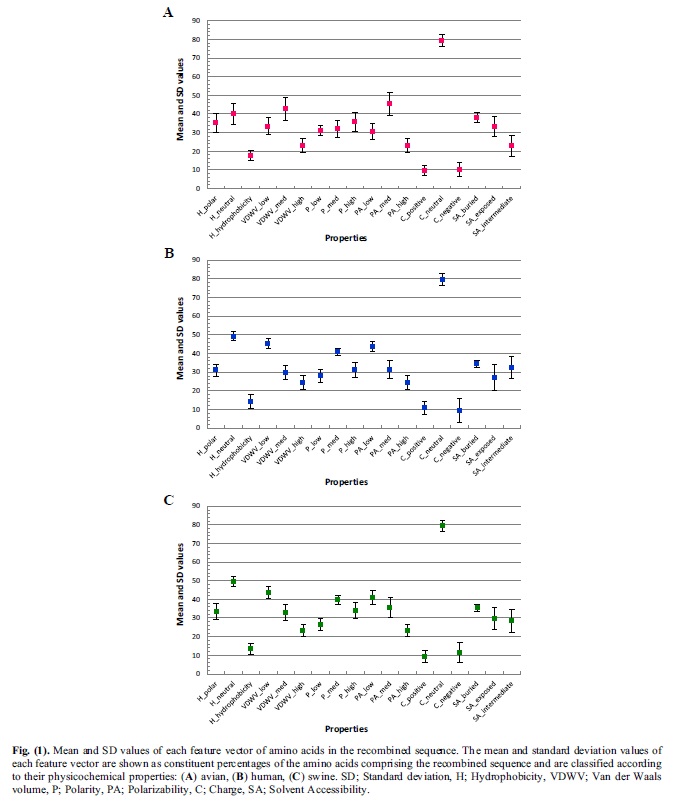
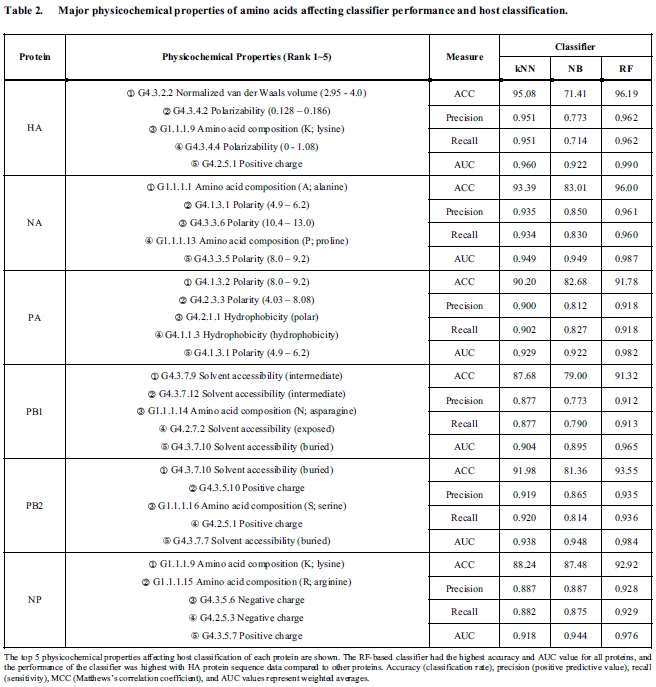
세 가지 알고리즘 중 kNN을 사용한 분류 정확도가 LT-Ag (ACC = 98.83)와 VP1 (ACC = 96.51) 모두에서 가장 높은 수치를 기록하였다. LT-Ag에서 숙주 분류와 가장 강한 상관관계를 보이는 아미노산 물리화학적 특성으로는 전하, 용매 접근성, 극성, 소수성 순인 것으로 나타났다. 그러나 VP1에서는 아미노산 조성이 숙주 분류와 가장 높은 상관관계를 가지는 것으로 나타났고, 그 뒤를 이어 전하, 정규화된 반 데르 발스 부피, 용매 접근성 순으로 확인되었다.
연구결론
본 연구의 결과는 다양한 숙주 종 사이에서 각기 다른 감염 특성을 나타내는 활성 및 신흥 PyV의 숙주 종을 전산학적으로 식별함으로써 분자 수준에서 PyV의 숙주 범위 및 감염 특성을 결정하거나 예측할 가능성이 있음을 시사한다. 이러한 아미노산 특성을 반영하는 숙주 종에서 LT-Ag 및 VP1 단백질의 구조 및 생화학적 차이는 PyV의 숙주 특이성을 결정하는 주요 요인으로 간주될 수 있을 것이라 제안될 수 있다.