Analysis of evolutionary and genetic patterns in structural genes of primate lentiviruses
GENES AND GENOMICS (2022)
.png)
연구배경
HIV는 유전적 차이가 큰 HIV1과 HIV2 두 종으로 나뉜다. 각기 다른 영장류 종(species)에 감염된 SIV로부터 유래된 것으로, HIV1과 HIV2는 감염의 지리적 분포나 질병 양상이 다르게 나타난다. 이 연구는 영장류 렌티바이러스의 전반적인 코돈 사용 패턴을 확인하고 HIV1, HIV2 및 SIV에서 공통적으로 또는 특이적으로 발현되는 진화적 및 유전적 특성을 탐구하는 것을 주요 목적으로 설계 및 수행되었다.
연구방법
HIV1, HIV2, SIV의 세가지 구조유전자 gag, pol, env의 염기 서열을 대상으로 계통 분석을 이용한 근연 관계 확인과 다양한 코돈 사용 인덱스로 코돈 사용 패턴, 중립성 진화 분석, 돌연변이 압력과 자연 선택 압력의 상대적 크기 평가, 인간의 코돈 사용에 대한 적응도를 확인하였다. 또한 계통수 및 종합적인 코돈 사용 패턴 분석으로 종간전파 우려가 높은 SIV 아종을 예측하였다.
연구결과

Gag, pol, env 유전자별 계통수 구축 결과 HIV1과 HIV2 모두 각각의 기원으로 밝혀진 SIV 종들과 함께 클러스터링된 양상을 확인할 수 있었다. 즉, 세 유전자 모두 HIV1는 SIVcpz 및 SIVgor와, HIV2는 SIVsm 및 SIVmacaca와 같은 분기군을 형성하였다. 또한 계통분류학적으로 숙주 원숭이 종들이 밀접하게 관련된 대부분의 경우 즉, 같은 속(genus)에 해당되면 감염된 SIV도 가까운 근연관계를 보임을 확인하였다.
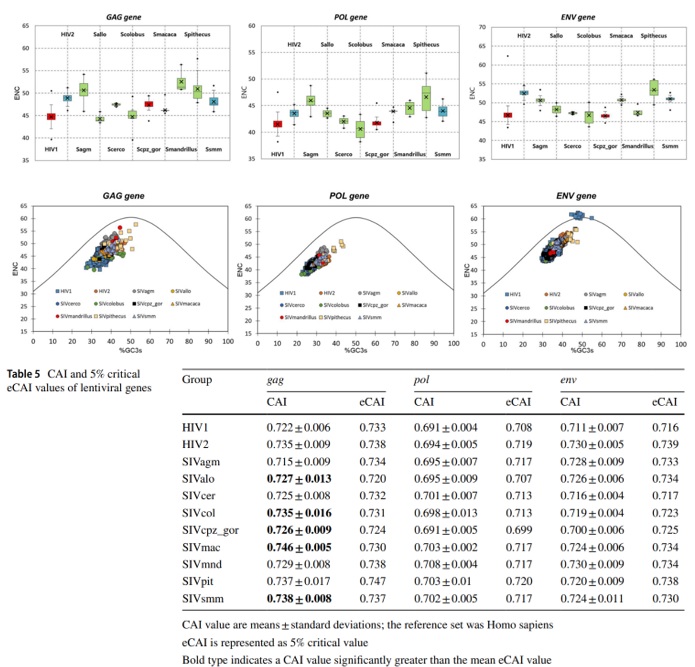
HIV1의 경우 HIV2에 비해 높은 코돈 사용 편향을 보이고 더 높은 선택압력을 견뎌내고 있는 것으로 나타났지만, 인간과의 코돈 사용 유사성은 HIV2가 오히려 더 높았다. 이를 통해 번역 효율이 HIV의 병원성 및 혈중 부하에 영향을 주는 주요한 요소가 아니라는 것이 확인되었다. 그리고 핵산 조성, 코돈 사용 편향, 선택압력의 크기, 숙주와의 코돈 사용 유사성 등에 대한 종합적인 분석으로 SIVcolobus 그룹과 SIVallo 그룹이 종간전파 우려가 높은 그룹으로 추정되었다.
연구결론
Gag 유전자 영역에서 HIV의 기원 바이러스인 SIVcpz_gor 및 SIVsmm 전체가 인간 tRNA pool과 보인 높은 유사성은 종간 이동이 성공적으로 이루어지기 위해서 gag 단백질의 빠른 합성이 필요하다는 것을 시사한다. 해당 연구 결과는 다양한 미생물에 대한 면역화를 위한 HIV 벡터 코돈 최적화의 참고 자료로 활용될 수 있을 뿐만 아니라 종간 이동으로 인한 새로운 HIV 종의 출현 가능성을 제기하여 이에 미리 대비할 수 있도록 유용 정보를 제공하였다는 데 의의가 있다.